
平台将构建不同长度的插入片段文库和短序列、双末端测序相结合的策略进行高通量测序,帮助客户在全基因组水平上扫描并检测与重要性状相关的基因序列差异和结构变异,实现遗传进化分析及重要性状候选基因预测,具有重大的科研和产业价值。
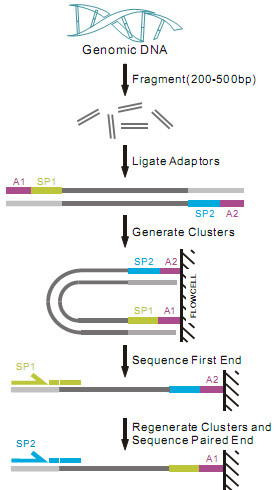
技术路线
服务内容
1. 测序
提取基因组DNA,进行片段化,电泳回收所需长度的DNA片段(0.2~0.5Kb),加上接头, 进行cluster制备,最后利用Paired-End(Solexa)的方法对插入片段进行重测序。
2. 数据处理
测序完成后,经过滤接头等污染后获得的reads,比对到参考基因组上,进行测序深度、覆盖度及均一性等产出数据的质量评估。
测序深度(Sequencing Depth)
测序得到的碱基总量(bp)与基因组大小(Genome)的比值,它是评价测序量的指标之一。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是Paired-End或Mate-Pair方案,当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证。
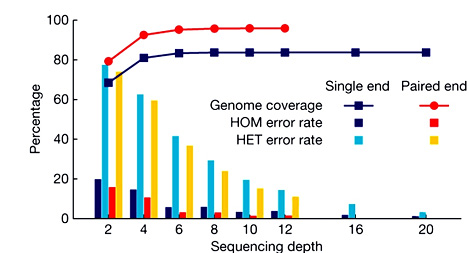
测序深度对基因组覆盖度和测序错误率的影响(HOM:纯合体 HET:杂合体)

